What is a phylogeny?
Phylogenies show the evolutionary relationships among a series of organisms, which can be graphically represented by phylogenetic trees. Phylogenetic trees are created to show relationships, but a more specific diagram that uses branch length to show evolutionary distance is called a phylogram. Both of these visual representations are created by comparing sequence data from DNA, RNA or proteins, and can either be based on the entire genome of organisms, or can be created based on specific short sequences, such as a single protein or DNA regions [1].
Sequence Alignment
How do you perform an alignment?
A sequence alignment allows you to compare multiple sequences to infer homology and their evolutionary relationships. Clustal Omega, MUSCLE, and T-Coffee are some of the many Multiple Sequence Alignment (MSA) programs available to compare three or more protein or nucleic acid sequences [2]. By inputting a series of FASTA codes, these programs use algorithms and statistical analyses to identify the best alignment method for a series of sequences. To statistically identify a best-fit sequence alignment, you can either use percent identity (PID) or BLOSUM62. PID takes into account the number of actual matches in your sequences, whereas BLOSUM analysis involves a matrix that creates a likelihood score for the event that an amino acid switch would occur between alignments. Phylogenetic Trees for Homo sapiens Pyrin isoform 1 using Clustal Omega |

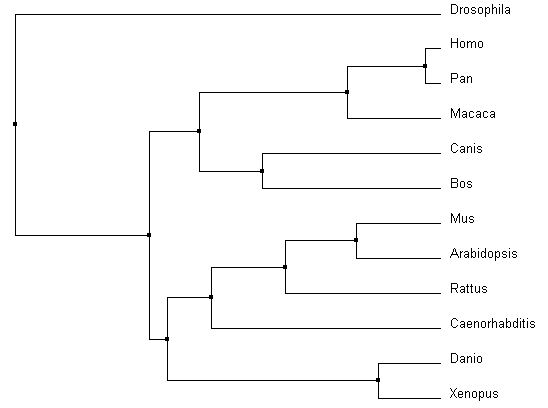
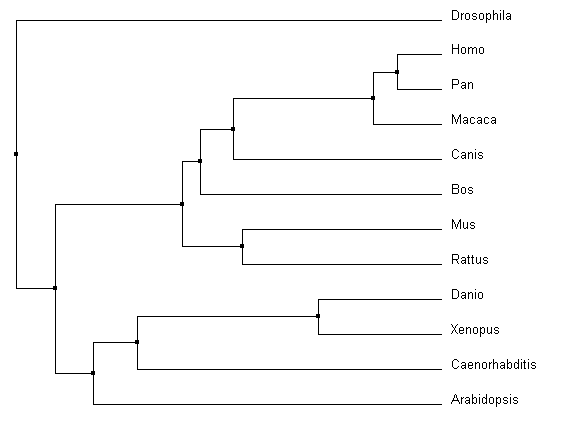
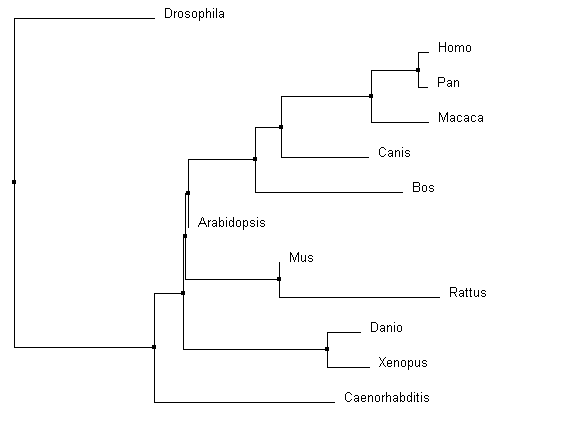
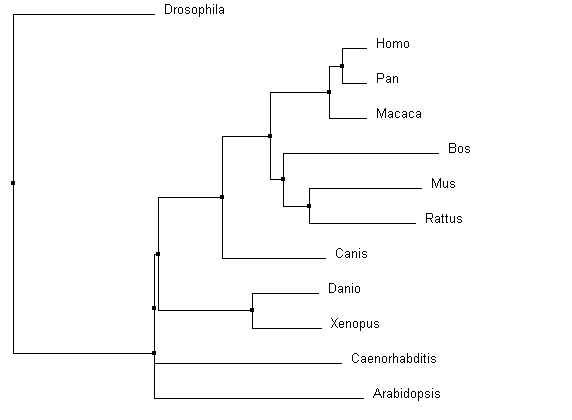
|
Analysis
First, it is important to realize that creating a phylogenetic tree from a single protein sequence is more limiting to describe evolutionary relationships than using a larger number of random proteins. This limitation can be compromised by creating phylogenetic trees using a number of different alignment tools and statistical tools to find a "best fit" sequence alignment. Furthermore, evolutionary relationships can be analyzed with known domain homology and Max ID from BLAST to identify the most accurate tree.
Similarities depicted between trees can be confirmed through domain homology and Max ID. All methods used identified Drosophila as the outgroup; identified the same core group of primates, Bos and Canis; and grouped Danio and Xenopus as similar. With the smallest Max ID with human pyrin and no common domains identified, Drosophila was accurately identified as the outgroup. The organisms consistently grouped in the core of the tree all have the greatest Max ID (over 50%) and all contained the same MEFV (TRIM20) domain structure. Danio BTY and Xenopus TRIM39 are 89% similar according to BLAST, and have the same TRIM domain structures. These commonalities were found in all the alignments. However, the most accurate alignments can be identified through more subtle relationship changes. PID in ClustalOmega (Figure 3 and 4) depicted Arabidopsis amidst Mus and Rattus in protein sequence. The protein in Arabidopsis was not shown to have any identifiable domains, whereas Mus and Rattus were shown to conserve two domains from the human pyrin. The Average distance model also removed Danio and Xenopus from the other vertebrates, despite the fact they both are harboring a similar TRIM protein. The neighbor joining tree using BLOSUM62 algorithm was the most accurate relative to prior knowledge about homology, as it co-localized all the organisms with known TRIM proteins. |
Comparison with Other Alignment Programs
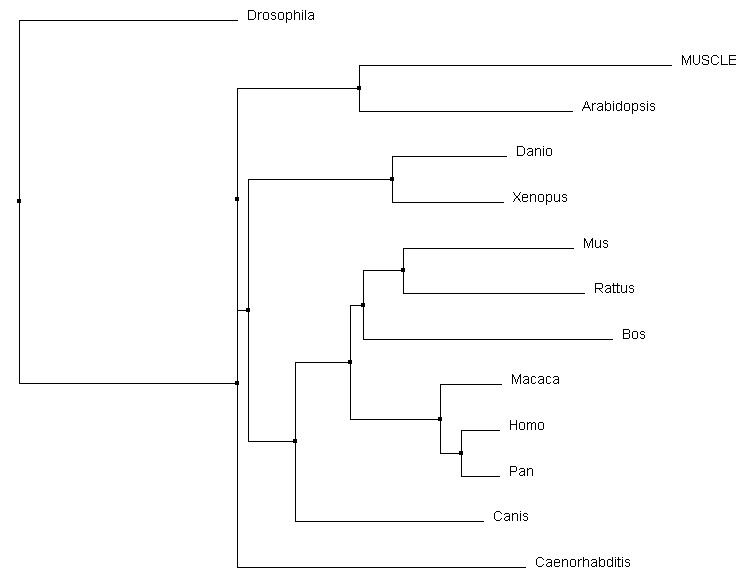
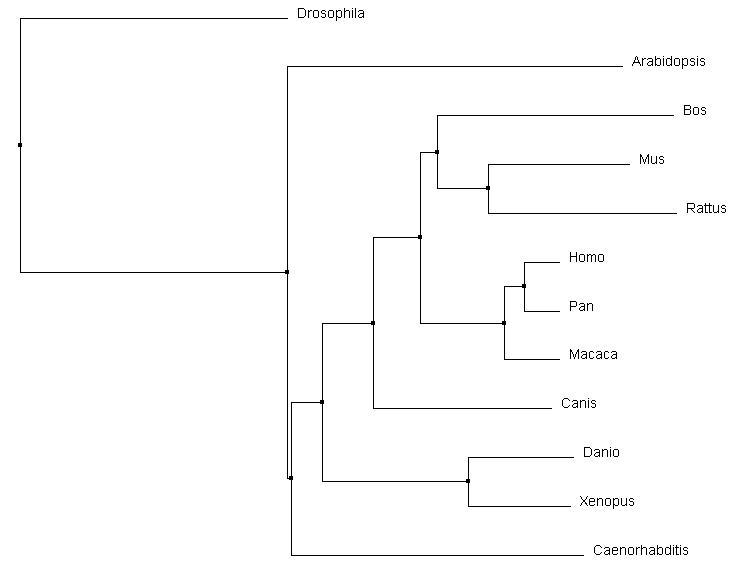
|
In comparing Clustal Omega with MUSCLE and T-coffee, the changes are very subtle. The same relationships were acknowledged for all of the vertebrates, including that of Canis and Bos in relation to Mus and Rattus. However, there were slight rearrangements can be identified between Caenorhabditis, Arabidopsis, and Drosophila and their connection to the core vertebrates. Overall, all three alignment tools produced similar results for pyrin and it's homologs.
|
FASTA sequences for homologs of Homo sapiens Pyrin isoform 1
FASTA sequences used for comparison are elaborated more fully on the Protein Homology page.
| pyrin_homology.txt |
